

ATOMISTIC LEVEL STUDIES OF SURFACES AND INTERFACES USING COMPUTATIONAL TECHNIQUES
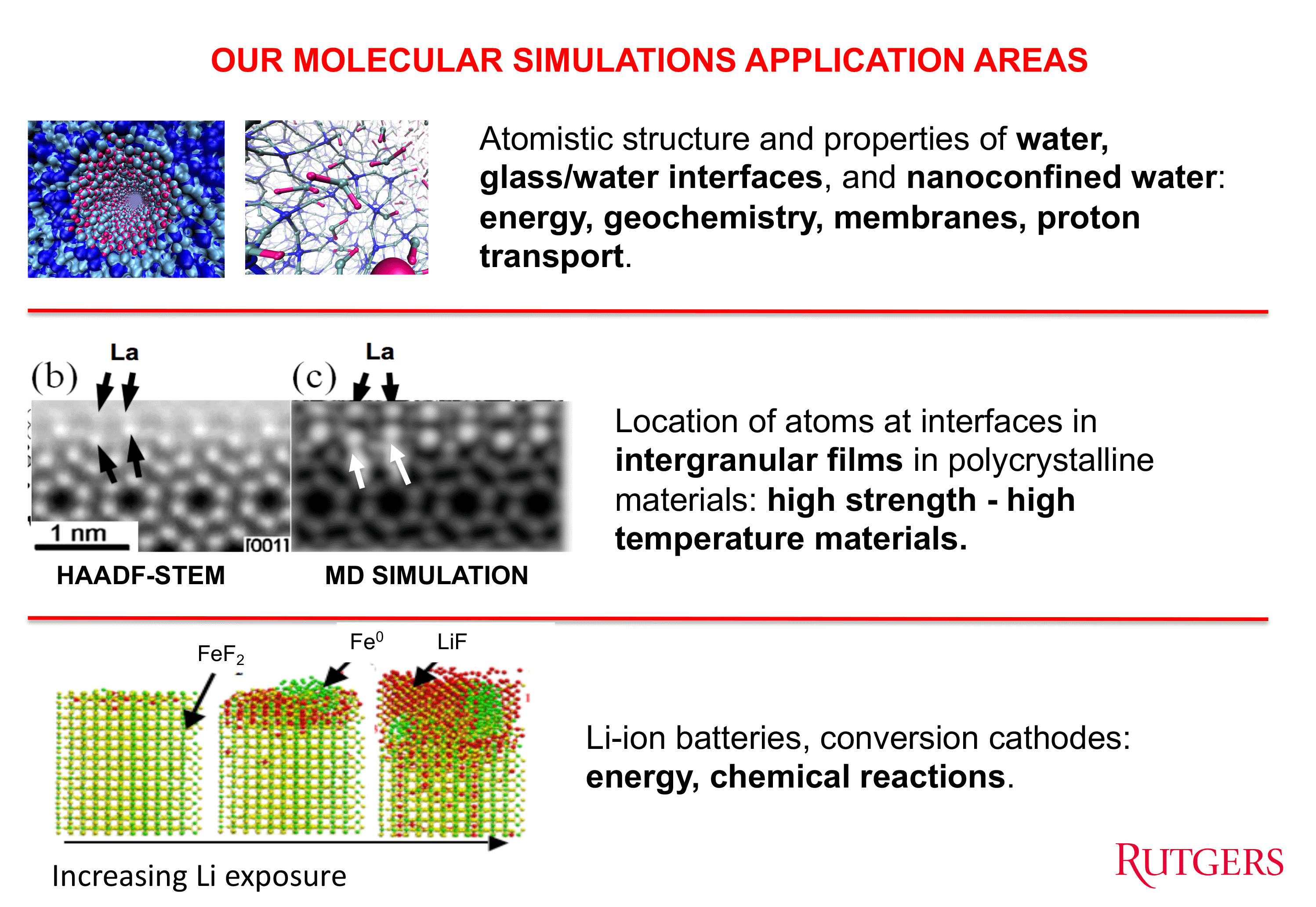
We are using molecular dynamics computer simulations to study the interfaces in materials. We have developed highly robust reactive interatomic potentials that provide atomistic behavior that is consistent with experimental studies and/or ab-initio calculations. We have then used such potentials to predict atomistic behavior that was subsequently verified by the most modern experimental techinques, such as HAADF-STEM, EELS, EXAFS, NMR, etc.
RECENT RESEARCH TOPICS
- Water/glass interfacial reactions
- Hydrogen bond lifetimes in bulk water 2013, versus at the water/glass interface and implications on proton transfer 2014
- Hydrogen bonding, proton transfer, auto-dissociation in bulk water (PhysChemChemPhys 2018 HOT paper)
- Effect of the silica surface on hydrogen bond lifetimes and proton transfer 2019
- Effect of moisture in silica glass on volume changes of the glass
- Effect of the presence of the water/silica interface on the structure and expansion of nanoconfined water: ChemPhysChem2008, Langmuir2009
- Atomistic structure of multicomponent silicate glasses and surfaces
- Atomistic structure of optical fibers used for very high energy applications
- Effect of nanocrystal surface orientation on phase changes during Li ion insertion in conversion batteries